用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
原料技术
›
体外诊断试剂主要原材料研究资料-人类 BCR/ABL融合基因P ...
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
6192
|
回复:
0
[分享]
体外诊断试剂主要原材料研究资料-人类 BCR/ABL融合基因P210 RNA扩增检测试剂盒(数字 PCR 法)
[复制链接]
青草
青草
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
发表于 2025-3-18 05:19
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
人类
BCR/ABL融合基因P210 RNA扩增检测试剂盒(数字
PCR 法)主要原材料研究资料
主要原材料研究资料
提交申报产品主要原材料的研究资料,内容包括主要原材料的来源、选择、制备方法的研究资料及其质量标准的制订资料、评价结果和质量分析证书,并详细描述原材料的技术指标和验收标准。
其他医疗器械/体外诊断试剂注册资料模板参见:
【墨斗鱼平台】注册资料分享
一、概述
白血病是我国十大高发恶性肿瘤之一,近年的临床研究表明,大部分白血病和淋巴瘤存在染色体畸变,如易位、缺失和突变等。染色体易位畸变大部分情况下会形成相关的融合基因,是白血病和淋巴瘤临床诊断的重要指标之一。
由t(9;22)(q34;q11)产生的费城染色体(Ph)在血液肿瘤中具有重要的诊断和预后意义,出现于90%以上的CML、30%成人ALL、2%~20%儿童ALL以及少数AML和MM患者。位于9q34的ABL基因与位于22q11的BCR基因相互易位产生的BCR-ABL融合基因会编码一种癌蛋白(p210BCR-ABL),95%的CML患者表现为P210阳性,在ph阳性ALL中,P210约占1/3,因此融合基因BCR-ABL的亚型P210可作为白血病分型诊断、疗效评价以及治疗监测的可靠指标。
本报告主要是关于人类BCR/ABL融合基因P210RNA扩增检测试剂盒(数字PCR法)主要原材料的研究,包括该试剂盒的相关信息以及主要原料的性状、性能等的研究。
二、人类
BCR/ABL融合基因P210 RNA扩增检测试剂盒(数字PCR法)检测原理及相关信息
2.1检测原理
使用随机引物反转录结合液滴数字PCR(ddPCR)技术进行定量检测。e13a2(b2a2)和e14a2(b3a2)BCR-ABL易位同时扩增、检测和量化。样本为人类血液中提取的总RNA,对FAM通道中的两个BCR-ABL融合转录本e13a2(b2a2)和/或e14a2(b3a2)的副本进行定量检测,并以ABL转录本内源性对照进行定量。将BCR-ABL转录本的数量量化为BCR-ABL/ABL的比率,然后返回国际范围内的值。
2.2人类
BCR/ABL融合基因P210 RNA扩增检测试剂盒(数字PCR法)
产品信息
包装规格:48人份/盒。
储存条件:-20℃条件下避光保存,有效期12个月。拆封后未使用完的试剂请立即-20℃条件下避光保存,并在8周内使用完且反复冻融次数不超过8次。
2.3人类
BCR/ABL融合基因P210 RNA扩增检测试剂盒(数字PCR法)
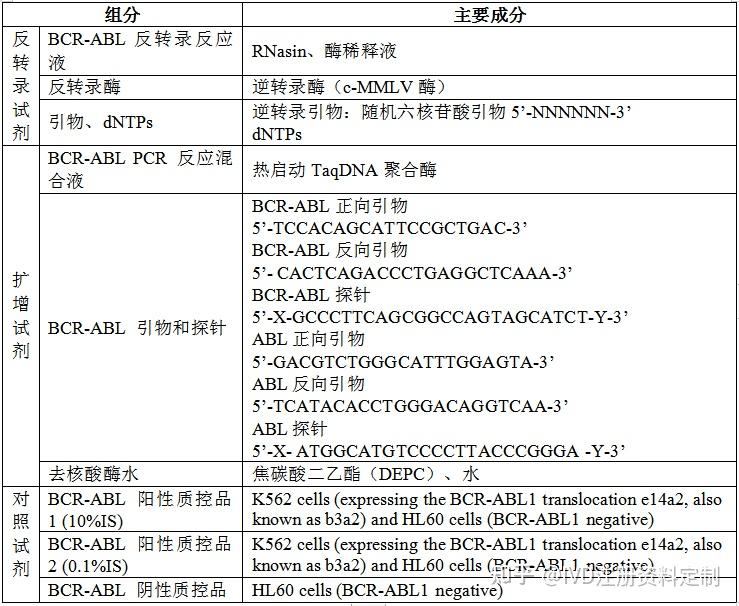
组分
三、主要原料来源及制备方法
3.1 RNasin:将转染有包含RNasin表达基因的BL21(DE3)大肠杆菌于LB培养基中进行诱导表达;然后将大肠杆菌裂解产物用RNase A亲和层析进行纯化制得。
3.2逆转录酶(c-MMLV酶):将逆转录酶基因插入至载体pET28a上,产生pET28a-MMLV RT质粒,具体合成序列包括3’端添加的纯化标签6×His序列和TAA终止密码子。将此质粒转化大肠杆菌BL21感受态细胞进行表达宿主构建,该质粒诱导表达后产生C端融合6×His纯化标签的MMLV逆转录酶。取发酵菌体以Ni亲和层析柱结合缓冲液重悬后,以细胞高压匀浆破碎仪破碎,低温高速离心收集破碎上清液,4℃环境下使用Ni亲和层析纯化柱进行纯化收集,得到MMLV逆转录酶突变体,配制于存储缓冲液中,低温保存。
3.3热启动TaqDNA聚合酶:
根据GenBank公布的Taq DNA聚合酶基因序列D32013 .1,按照大肠杆菌偏好密码子,人工合成优化的Taq DNA聚合酶基因片段;然后将优化的Taq DNA聚合酶基因片段构建到原核表达载体上,进行大肠杆菌转化和筛选,获得表达Taq DNA聚合酶的重组菌;然后依次进行诱导表达、细胞破壁、热变性、梯度盐析、透析和层析,完成Taq DNA聚合酶的浓缩和纯化;最后,将抗Taq DNA聚合酶单克隆抗体与Taq DNA聚合酶进行特异性结合,完成Taq DNA聚合酶的配基修饰,即为热启动Taq DNA聚合酶。
3.4DNA引物和探针:使用****核酸分析软件分别对已知的融合基因BCR-ABL(P210)基因的核苷酸序列进行同源性比较,在找到同源区段的基础上,进一步使用****引物设计软件选择并设计寡核苷酸引物和探针。由于所设计的引物和探针均具有互补于BCR-ABL(P210)基因的序列,而且与其他病原体的核苷酸序列没有同源性,也不包括任何常见的核酸内切酶的酶切位点,所以避免了融合基因BCR-ABL(P210)检测的假阴性和假阳性,提高了检测的可靠性和准确度。使用DNA合成装置合成所需的寡核苷酸引物和探针,用分子筛和快速蛋白质液相层析法(FPLC)纯化后进行氨解处理。同样合成所需的探针序列,氨解处理后分别在其5’端标记作为荧光发生基团(报告基团)的6-FAM amidite,并在其3’端标记借助活性连接臂偶联上作为荧光淬灭或抑制基团的BHQ1。以FPLC层析法纯化荧光标记的探针。然后,使用变性条件下的聚丙烯酰胺凝胶(20%)电泳法和分光光度法物理鉴定所合成的引物和探针。
经检索,BCR/ABL mRNA (e13a2)序列如下:
gcgaacaagggcagcaaggctacggagaggctgaagaagaagctgtcggagcaggagtcactgctgctgcttatgtctcccagcatggccttcagggtgcacagccgcaacggcaagagttacacgttcctgatctcctctgactatgagcgtgcagagtggagggagaacatccgggagcagcagaagaagtgtttcagaagcttctccctgacatccgtggagctgcagatgctgaccaactcgtgtgtgaaactccagactgtccacagcattccgctgaccatcaataaggaagaa
gcccttcagcggccagtagcatct
gactttgagcctcagggtctgagtgaagccgctcgttggaactccaaggaaaaccttctcgctggacccagtgaaaatgaccccaaccttttcgttgcactgtatgattttgtggccagtggagataacactctaagcataactaaaggtgaaaagctccgggtcttaggctataatcacaatggggaatggtgtgaagcccaaaccaaaaatggccaaggctgggtcccaagcaactacatcacgccagtcaacagtctggagaaacactcctggtaccatgggcctgtgtcccgcaatgccgctgagtatctgctgagcagcgggatcaatggcagcttcttggtgcgtgagagtgagagcagtcctggccagaggtccatctcgctgagatacgaagggagggtgtaccattacaggatcaacactgcttctgatggcaagctctacgtctcctccgagagccgcttcaacaccctggccgagttggttcatcatcattcaacggtggccgacgggctcatcaccacgctccattatccagccccaaagcgcaacaagcccactgtctatggtgtgtcccccaactacgacaagt
BCR/ABL mRNA (e14a2)序列如下:gcgaacaagggcagcaaagctacggagaggctgaagaagaagctgtcggagcaggagtcactgctgctgcttatgtctcccagcatggccttcagggtgcacagccgcaacggcaagagttacacgttcctgatctcctctgactatgagcgtgcagagtggagggagaacatccgggagcagcagaagaagtgtttcagaagcttctccctgacatccgtggagctgcagatgctgaccaactcgtgtgtgaaactccagactgtccacagcattccgctgaccatcaataaggaagatgatgagtctccggggctctatgggtttctgaatgtcatcgtccactcagccactggatttaagcagagttcaaaa
gcccttcagcggccagtagcatct
gactttgagcctcagggtctgagtgaagccgctcgttggaactccaaggaaaaccttctcgctggacccagtgaaaatgaccccaaccttttcgttgcactgtatgattttgtggccagtggagataacactctaagcataactaaaggtgaaaagctccgggtcttaggctataatcacaatggggaatggtgtgaagcccaaaccaaaaatggccaaggctgggtcccaagcaactacatcacgccagtcaacagtctggagaaacactcctggtaccatgggcctgtgtcccgcaatgccgctgagtatctgctgagcgcgggatcaatggcagcttcttggtgcgtgagagtgagagcagtcctggccagaggtccatctcgctgagatacgaagggagggtgtaccattacaggatcaacactgcttctgatggcaagctctacgtctcctccgagagccgcttcaacaccctggccgagttggttcatcatcattcaacggtggccgacgggctcatcaccacgctccattatccagccccaaagcgcaacaagcccactgtctatggtgtgtcccctaactacgacaagt
e13a2、e14a2引物和探针序列一致。
BCR-ABL正向引物序列:tccacagcattccgctgac
BCR-ABL反向引物序列位置:tttgagcctcagggtctgagtg
BCR-ABL探针序列:
gcccttcagcggccagtagcatct
tgatgagtctccggggctctatgggtttctgaatgtcatcgtccactcagccactggatttaagcagagttcaaa为e14a2比e13a2多出部分。
内参基因ABL引物和探针的设计原则同上,5’端标记荧光发生基团(报告基团)为HEX。
经检索,ABL mRNA序列如下:
gcgaacaagggcagcaaggctacggagaggctgaagaagaagctgtcggagcaggagtcactgctgctgcttatgtctcccagcatggccttcagggtgcacagccgcaacggcaagagttacacgttcctgatctcctctgactatgagcgtgcagagtggagggagaacatccgggagcagcagaagaagtgtttcagaagcttctccctgacatccgtggagctgcagatgctgaccaactcgtgtgtgaaactccagactg
tccacagcattccgctgac
catcaataaggaagaa
gcccttcagcggccagtagcatct
gac
tttgagcctcagggtctgagtg
aagccgctcgttggaactccaaggaaaaccttctcgctggacccagtgaaaatgaccccaaccttttcgttgcactgtatgattttgtggccagtggagataacactctaagcataactaaaggtgaaaagctccgggtcttaggctataatcacaatggggaatggtgtgaagcccaaaccaaaaatggccaaggctgggtcccaagcaactacatcacgccagtcaacagtctggagaaacactcctggtaccatgggcctgtgtcccgcaatgccgctgagtatctgctgagcagcgggatcaatggcagcttcttggtgcgtgagagtgagagcagtcctggccagaggtccatctcgctgagatacgaagggagggtgtaccattacaggatcaacactgcttctgatggcaagctctacgtctcctccgagagccgcttcaacaccctggccgagttggttcatcatcattcaacggtggccgacgggctcatcaccacgctccattatccagccccaaagcgcaacaagcccactgtctatggtgtgtcccccaactacgacaagt
BCR/ABL mRNA (e14a2)序列如下:gcgaacaagggcagcaaagctacggagaggctgaagaagaagctgtcggagcaggagtcactgctgctgcttatgtctcccagcatggccttcagggtgcacagccgcaacggcaagagttacacgttcctgatctcctctgactatgagcgtgcagagtggagggagaacatccgggagcagcagaagaagtgtttcagaagcttctccctgacatccgtggagctgcagatgctgaccaactcgtgtgtgaaactccagactg
tccacagcattccgctgac
catcaataaggaaga
tgatgagtctccggggctctatgggtttctgaatgtcatcgtccactcagccactggatttaagcagagttcaaa
a
gcccttcagcggccagtagcatct
gac
tttgagcctcagggtctgagtg
aagccgctcgttggaactccaaggaaaaccttctcgctggacccagtgaaaatgaccccaaccttttcgttgcactgtatgattttgtggccagtggagataacactctaagcataactaaaggtgaaaagctccgggtcttaggctataatcacaatggggaatggtgtgaagcccaaaccaaaaatggccaaggctgggtcccaagcaactacatcacgccagtcaacagtctggagaaacactcctggtaccatgggcctgtgtcccgcaatgccgctgagtatctgctgagcgcgggatcaatggcagcttcttggtgcgtgagagtgagagcagtcctggccagaggtccatctcgctgagatacgaagggagggtgtaccattacaggatcaacactgcttctgatggcaagctctacgtctcctccgagagccgcttcaacaccctggccgagttggttcatcatcattcaacggtggccgacgggctcatcaccacgctccattatccagccccaaagcgcaacaagcccactgtctatggtgtgtcccctaactacgacaagt
e13a2、e14a2引物和探针序列一致。
BCR-ABL正向引物序列:tccacagcattccgctgac
BCR-ABL反向引物序列位置:tttgagcctcagggtctgagtg
BCR-ABL探针序列:
gcccttcagcggccagtagcatct
tgatgagtctccggggctctatgggtttctgaatgtcatcgtccactcagccactggatttaagcagagttcaaa
为e14a2比e13a2多出部分。
内参基因ABL引物和探针的设计原则同上,5’端标记荧光发生基团(报告基团)为HEX。
经检索,ABL mRNA序列如下:
gttaacaggcgcgtcccggccaggcggagacgcggccgcggccatgggcgggcgcgggcgcgcggggcggcggtgagggcggctggcggggccgggggcgccgggggggcgcgcgggccgagccgggcctgagccgggcccgcggaccgagctgggagaggggttccggcccccgacgtgctggcgcgggaaaatgttggagatctgcctgaagctggtgggctgcaaatccaagaaggggctgtcctcgtcctccagctgttatctggaagaagcccttcagcggccagtagcatctgac
tttgagcctcagggtctgagtg
aagccgctcgttggaactccaaggaaaaccttctcgctggacccagtgaaaatgaccccaaccttttcgttgcactgtatgattttgtggccagtggagataacactctaagcataactaaaggtgaaaagctccgggtcttaggctataatcacaatggggaatggtgtgaagcccaaaccaaaaatggccaaggctgggtcccaagcaactacatcacgccagtcaacagtctggagaaacactcctggtaccatgggcctgtgtcccgcaatgccgctgagtatctgctgagcagcgggatcaatggcagcttcttggtgcgtgagagtgagagcagtcctggccagaggtccatctcgctgagatacgaagggagggtgtaccattacaggatcaacactgcttctgatggcaagctctacgtctcctccgagagccgcttcaacaccctggccgagttggttcatcatcattcaacggtggccgacgggctcatcaccacgctccattatccagccccaaagcgcaacaagcccactgtctatggtgtgtcccccaactacgacaagtgggagatggaacgcacggacatcaccatgaagcacaagctgggcgggggccagtacggggaggtgtacgagggcgtgtggaagaaatacagcctgacggtggccgtgaagaccttgaaggaggacaccatggaggtggaagagttcttgaaagaagctgcagtcatgaaagagatcaaacaccctaacctggtgcagctccttggggtctgcacccgggagcccccgttctatatcatcactgagttcatgacctacgggaacctcctggactacctgagggagtgcaaccggcaggaggtgaacgccgtggtgctgctgtacatggccactcagatctcgtcagccatggagtacctggagaagaaaaacttcatccacagagatcttgctgcccgaaactgcctggtaggggagaaccacttggtgaaggtagctgattttggcctgagcaggttgatgacaggggacacctacacagcccatgctggagccaagttccccatcaaatggactgcacccgagagcctggcctacaacaagttctccatcaagtccgacgtctgggcatttggagtattgctttgggaaattgctacct
atggcatgtccccttacccggga
attgacctgtcccaggtgtatgagctgctagagaaggactaccgcatggagcgcccagaaggctgcccagagaaggtctatgaactcatgcgagcatgttggcagtggaatccctctgaccggccctcctttgctgaaatccaccaagcctttgaaacaatgttccaggaatccagtatctcagacgaagtggaaaaggagctggggaaacaaggcgtccgtggggctgtgagtaccttgctgcaggccccagagctgcccaccaagacgaggacctccaggagagctgcagagcacagagacaccactgacgtgcctgagatgcctcactccaagggccagggagagagcgatcctctggaccatgagcctgccgtgtctccattgctccctcgaaaagagcgaggtcccccggagggcggcctgaatgaagatgagcgccttctccccaaagacaaaaagaccaacttgttcagcgccttgatcaagaagaagaagaagacagccccaacccctcccaaacgcagcagctccttccgggagatggacggccagccggagcgcagaggggccggcgaggaagagggccgagacatcagcaacggggcactggctttcacccccttggacacagctgacccagccaagtccccaaagcccagcaatggggctggggtccccaatggagccctccgggagtccgggggctcaggcttccggtctccccacctgtggaagaagtccagcacgctgaccagcagccgcctagccaccggcgaggaggagggcggtggcagctccagcaagcgcttcctgcgctcttgctccgcctcctgcgttccccatggggccaaggacacggagtggaggtcagtcacgctgcctcgggacttgcagtccacgggaagacagtttgactcgtccacatttggagggcacaaaagtgagaagccggctctgcctcggaagagggcaggggagaacaggtctgaccaggtgacccgaggcacagtaacgcctccccccaggctggtgaaaaagaatgaggaagctgctgatgaggtcttcaaagacatcatggagtccagcccgggctccagcccgcccaacctgactccaaaacccctccggcggcaggtcaccgtggcccctgcctcgggcctcccccacaaggaagaagctggaaagggcagtgccttagggacccctgctgcagctgagccagtgacccccaccagcaaagcaggctcaggtgcaccagggggcaccagcaagggccccgccgaggagtccagagtgaggaggcacaagcactcctctgagtcgccagggagggacaaggggaaattgtccaggctcaaacctgccccgccgcccccaccagcagcctctgcagggaaggctggaggaaagccctcgcagagcccgagccaggaggcggccggggaggcagtcctgggcgcaaagacaaaagccacgagtctggttgatgctgtgaacagtgacgctgccaagcccagccagccgggagagggcctcaaaaagcccgtgctcccggccactccaaagccacagtccgccaagccgtcggggacccccatcagcccagcccccgttccctccacgttgccatcagcatcctcggccctggcaggggaccagccgtcttccaccgccttcatccctctcatatcaacccgagtgtctcttcggaaaacccgccagcctccagagcggatcgccagcggcgccatcaccaagggcgtggtcctggacagcaccgaggcgctgtgcctcgccatctctaggaactccgagcagatggccagccacagcgcagtgctggaggccggcaaaaacctctacacgttctgcgtgagctatgtggattccatccagcaaatgaggaacaagtttgccttccgagaggccatcaacaaactggagaataatctccgggagcttcagatctgcccggcgacagcaggcagtggtccagcggccactcaggacttcagcaagctcctcagttcggtgaaggaaatcagtgacatagtgcagaggtagcagcagtcaggggtcaggtgtcaggcccgtcggagctgcctgcagcacatgcgggctcgcccatacccgtgacagtggctgacaagggactagtgagtcagcaccttggcccaggagctctgcgccaggcagagctgagggccctgtggagtccagctctactacctacgtttgcaccgcctgccctcccgcaccttcctcctccccgctccgtctctgtcctcgaattttatctgtggagttcctgctccgtggactgcagtcggcatgccaggacccgccagccccgctcccacctagtgccccagactgagctctccaggccaggtgggaacggctgatgtggactgtctttttcatttttttctctctggagcccctcctcccccggctgggcctccttcttccacttctccaagaatggaagcctgaactgaggccttgtgtgtcaggccctctgcctgcactccctggccttgcccgtcgtgtgctgaagacatgtttcaagaaccgcatttcgggaagggcatgcacgggcatgcacacggctggtcactctgccctctgctgctgcccggggtggggtgcactcgccatttcctcacgtgcaggacagctcttgatttgggtggaaaacagggtgctaaagccaaccagcctttgggtcctgggcaggtgggagctgaaaaggatcgaggcatggggcatgtcctttccatctgtccacatccccagagcccagctcttgctctcttgtgacgtgcactgtgaatcctggcaagaaagcttgagtctcaagggtggcaggtcactgtcactgccgacatccctcccccagcagaatggaggcaggggacaagggaggcagtggctagtggggtgaacagctggtgccaaatagccccagactgggcccaggcaggtctgcaagggcccagagtgaaccgtcctttcacacatctgggtgccctgaaagggcccttcccctcccccactcctctaagacaaagtagattcttacaaggccctttcctttggaacaagacagccttcacttttctgagttcttgaagcatttcaaagccctgcctctgtgtagccgccctgagagagaatagagctgccactgggcacctgcgcacaggtgggaggaaagggcctggccagtcctggtcctggctgcactcttgaactgggcgaatgtcttatttaattaccgtgagtgacatagcctcatgttctgtgggggtcatcagggagggttaggaaaaccacaaacggagcccctgaaagcctcacgtatttcacagagcacgcctgccatcttctccccgaggctgccccaggccggagcccagatacgggggctgtgactctgggcagggacccggggtctcctggaccttgacagagcagctaactccgagagcagtgggcaggtggccgcccctgaggcttcacgccgggagaagccaccttcccaccccttcataccgcctcgtgccagcagcctcgcacaggccctagctttacgctcatcacctaaacttgtactttatttttctgatagaaatggtttcctctggatcgttttatgcggttcttacagcacatcacctctttgcccccgacggctgtgacgcagccggagggaggcactagtcaccgacagcggccttgaagacagagcaaagcgcccacccaggtcccccgactgcctgtctccatgaggtactggtcccttccttttgttaacgtgatgtgccactatattttacacgtatctcttggtatgcatcttttatagacgctcttttctaagtggcgtgtgcatagcgtcctgccctgccccctcgggggcctgtggtggctccccctctgcttctcggggtccagtgcattttgtttctgtatatgattctctgtggttttttttgaatccaaatctgtcctctgtagtattttttaaataaatcagtgtttacattagaa
其中ABL正向引物:5’-GACGTCTGGGCATTTGGAGTA-3’
ABL反向引物位置:5’-ttgacctgtcccaggtgtatga-3’
ABL探针:
5’-atggcatgtccccttacccggga-3’
3.5去核酸酶水:用超纯的去离子水经DEPC处理后,再经过高压灭菌而来。经检测无核酸酶和蛋白酶活性,可用于cDNA合成、体外转录、RNA提取等对核酸酶敏感的分子生物学试验。
3.6 K562细胞系:K-562是由Lozzio从一位临终的53岁女性慢性骨髓性白血病患者的胸水中建立。K-562细胞被归入高度未分化的粒性白细胞类,Erson等对其表面膜性质的研究表明:K-562细胞是一株人类红白血病细胞。它能表达BCR-ABL1 e14a2(b3a2) ,是WHO第一代BCR-ABL1融合基因标准物质NIBSC code: 09/138所使用的细胞株。通过细胞传代培养获得。
3.7 HL60 细胞系:BCR-ABL1 阴性细胞系,该细胞由Collins SJ从一位患有急性早幼粒细胞性白血病的36岁白人女性的外周血中分离建立。是WHO第一代BCR-ABL1融合基因标准物质NIBSC code: 09/138所使用的细胞株(用做内参)。通过细胞传代培养获得。
三、主要原料选择及质量标准制定资料
4.1逆转录酶(c-MMLV酶的选择与质量标准的制定)
4.1.1制定逆转录酶活性的检测方法
4.1.1.1实验仪器:
荧光PCR仪。
4.1.1.2试剂
4.1.1.2.1 发夹型寡核苷酸序列(T2),序列为:
5'-tagcgaaggatgtgaacctaatcecTGCTCCCGCGGCCGatctgcCGGCCGCGGGAGCA-3'
4.1.1.2.2 dNTP。
注:dGTP:三磷酸鸟嘌呤脱氧核苷酸;dATP:三磷酸腺嘌呤脱氧核苷酸;dTTP:三磷酸胸腺嘧啶脱氧核苷酸;dCTP:三磷酸胞嘧啶脱氧核苷酸,dNTP;三磷酸碱基脱氧核苷酸,包括 dGTP、dATP、dTTP、dCTP。包括dCTP、dATP、dTTP、dGTP。
4.1.1.2.3 荧光核酸染料。
4.1.1.2.4 Lamda DNA标准品。
4.1.1.3溶液的配制
(1)1×TE溶液:10 mmol/L Tris-HCl,1 mmol/L乙二胺四乙酸二钠溶液,pH 8.5(25 ℃)。
(2)PCR 缓冲液;500 mmol/LTris-HCl,750 mmol/L 氯化钾溶液,100 mmol/L DTT,pH 8.5 (25 ℃)。
(3)Lambda DNA预染液:以8.5 μL:1μL:0.5 μL的比例分别量取纯化水、10×PCR缓冲液和荧光核酸染料加入离心管,在漩涡混匀器上振荡混匀1 min 后在微型离心机上离心数秒,储存于2 ℃~8℃。
(4)氯化镁溶液:25 mmol/L。;
(5)酶稀释缓冲液:10 mmol/L Tris-HCl,0.1 mmol/L 乙二胺四乙酸二钠溶液,1 mmol/L DTT,50%(体积分数)甘油,pH8.0(25 ℃)。
(6) T2 溶液:将发夹型寡核苷酸序列(T2)干粉按照合成单上的说明稀释到 100 μmol/L,储存于-20 ℃。使用时从-20 ℃取出平衡至室温,于漩涡混匀器上混匀10 s,在微型离心机上离心数秒。
(7)dNTP混合液,将dNTP中的dCTP、dATP、dTTP、dGTP等体积进行温合,于漩涡混匀器上混匀10s,在微型离心机上离心数秒,配制成25 mmol/L的dNTP混合液。
(8)Lambda DNA标准品:使用1×TE溶液将Lambda DNA 标准品进行4倍连续梯度稀释,得到100μg/mL、25μg/mL、12.5 μg/mL、6.25 μg/mL、3.125 μg/mL、1.5625μg/mL和0.78125μg/mL的8个浓度梯度溶液,以相同体积的1×TE溶液作为背景对照。各梯度溶液和背景对照分别加入等体积的预染液,于漩涡混匀器上混匀10 s,在微型离心机上离心数秒,以 20 μL/管分装于PCR八联管内,每个梯度3个重复,盖紧管盖,在微型离心机上离心数秒后备用。
(9)M-MLV反转录酶梯度释溶液,根据标定的酶比活力,使用酶稀释缓冲液对M-MLV反转录酶进行适当稀释(如标定浓度为200 U/μL,则可2倍稀释,得到2×、4×的梯度稀释液)。该步骤需在冰上进行。
4.1.1.4试验步骤
4.1.1.4.1 聚合反应液的配制与分装
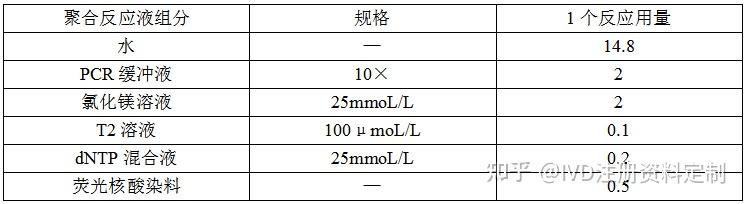
4.1.1.4.1.1 在微量离心管中按照表 1的比例配制聚合反应液(冰上配制),聚合反应液配制完成后,在漩涡混匀器涡旋混匀10 s,微型离心机离心数秒。
配制总体积计算见下式:
V=[3×(n+1)+1]×V1
式中:
V—总体积,单位为微升(μL);
3 —重复次数;
n—M-MLV反转录酶的稀释梯度数;
1—前一个1表示空白对照数;后一个1表示预留损失数量;
V—表示1个反应用量,见表1,单位为微升(μL)。
表1聚合反应液配比
4.1.1.4.1.2 将 4.1.5.1.1中的聚合反应液平均分装到 n 个(n为 M-MLV反转录酶的稀释梯度数)微量离心管中,分别加入1.2 μL 的各浓度梯度的 M-MLV反转录酶梯度稀释溶液和空白对照(酶稀释缓冲液),在漩涡混匀器涡旋混匀10 s,微型离心机离心数秒。各管混合液以 20 μL/管,分装于PCR八联管内,每管混合液3个重复,盖紧管盖,在微型离心机上离心数秒后备用。
4.1.1.4.2运行程序
在荧光 PCR仪上设置如下温度程序:
37℃ 16s
荧光采集通道:SYBR
120个循环
将装有Lambda DNA标准品和聚合反应液的 PCR八联管置干荧光 PCR.仪中.启动温康程序,开始聚合反应。
4.1.1.5数据分析
4.1.1.5.1 Lambda DNA标准曲线的制作
使用荧光 PCR仪的软件打开数据,导出文件,按以下步骤进行,按下式计算:
a)选取数据文件中 Lambda DNA 标准品和背景对照的第 25个循环的荧光值,并计算平均荧光值。
b) 将各梯度 Lambda DNA标准品的荧光值减掉背景对照的平均荧光值,得到净荧光值。
c) 计算各梯度净荧光值的平均值和标准偏差 SD。
d) 以Lambda DNA标准品 DNA量(ng)为 X 轴,以各梯度净荧光值的平均值为Y轴,标准偏差SD 为错误值进行多项式拟合分析,制作工作曲线,从中获得净荧光值与双链 DNA 浓度的关系曲线如下,R2≥0.98。
y=A+Bx
式中:
A —线性方程拟合曲线的截距;
B—线性方程拟合曲线的斜率;
C—y -荧光值;
x-—双链DNA的量,单位为纳克(ng)。
4.1.1.5.2 聚合反应新生成的双链 DNA量的计算
选取 Excel文件中待检 M-MLV反转录酶的第 70 个循环的荧光数据。首先计算空白对照3个重复的平均荧光值,将各梯度 M-MLV反转录酶的荧光值减掉空白对照的平均荧光值,得到净荧光值,计算各梯度净荧光值的平均值(y1)。将各净荧光值代人上式计算聚合反应新生成的双链 DNA量(x1)。
4.1.1.5.3 M-MLV反转录酶梯度条件下的 dNTP消耗量计算
M-MLV反转录酶梯度条件下的 dNTP消耗量以y2表示,单位为 nmol,按下式计算:
y2=x1/(2×324.5)
式中:
x1——新生成的双链DNA量,单位为纳克(ng);
2——双链 DNA由2条单链 DNA组成;
324.5-—dNTP的相对平均分子质量。
4.1.1.5.4 M-MLV反转录酶活力计算
根据M-MLV反转录酶活性单位定义,M-MLV反转录酶酶活力以S表示,单位为U/μL。选择在0.014 nmol~0.196 nmol 范围内的 dNTP消耗量按照下式计算:
S=(y2+0.045)×D/0.0012
式中:
y2——dNTP消耗量,单位为纳摩尔(nmol);
D——酶的稀释倍数。
4.1.2制定逆转录酶核酸外切酶活性的检测方法。
4.1.2.1 仪器和设备
4.1.2.1.1 恒温水浴槽。
4.1.2.1.2 台式离心机。
4.1.2.1.3 超净工作台。
4.1.2.1.4 恒温培养箱。
4.1.2.2 材料
4.1.2.2.1 高纯 CCC pUC19 DNA。
4.1.2.2.2 T4 DNA连接酶。
4.1.2.2.3 10× T4 DNA 连接酶缓冲液:500 mmol/L Tris-HCl 缓冲液(pH7.8),100 mmol/L Mg2+,10 mmol/L ATP,100 mmol/L DTT。
4.1.2.2.4 LB培养液。
4.1.2.2.5 氯仿-异戊醇(24:1,体积比)。
4.1.2.2.6 大肠杆菌DH5a(感受态)。
4.1.2.2.7 LB平板【含β-半乳糖苷酶,异丙基硫代半乳糖苷(IPTG),氨苄青霉素】。
4.1.2.3 实验步骤
4.1.2.3.1 长时程过量酶切
取10 μg 高纯 CCC pUC19 DNA 置于一只微量离心管中,加入 20 μL 10× BamHI缓冲液和150 μL水,充分混匀后平均分成A和B两管;A管加入5 μL BamHI(50 U~100 U)和5 μL水,达总体积100 μL;B管加人5μL BamHI(50 U~100 U)和5 μL M-MLV反转录酶(1 μg/μL)。混匀后置于37 ℃水浴槽,1 h后各取出 40 μL,其中 20 μL(标记 A1号和 B1号)待电泳检测,另 20 μL(标记A2号和 B2号)待连接及蓝白斑计数;所剩60 μL继续酶切达10 h 以上,又将它各均分为3管,分别标记A3、B3、A4、B4、A5、B5号。
4.1.2.3.2 电泳
4.1.2.3.2.1 取出 A1、B1、A3、B3号管,加入载样缓冲液,准备电泳检测。
4.1.2.3.2.2 1.5%琼脂糖,电泳电压5V/cm~10 V/cm,当溴酚蓝线到达凝胶板 2/3 时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照。
4.1.2.3.3 连接
4.1.2.3.3.1 回收 DNA
取出 A2、B2、A4、B4、A5、B5号管,各加入 20 μL氯仿-异戊醇先脱去杂蛋白,再沉淀出DNA,并真空干燥样品。
4.1.2.3.3.2 连接反应
每管加入2 μL10× T4 DNA连接酶缓冲液,2U T4 DNA连接酶,加入水达总体积 20 μL,混匀后置于15℃水浴槽8 h。
4.1.2.3.3.3 转化与铺板
4.1.2.3.3.3.1 将感受态菌液加入含连接处理过的 DNA 微量离心管内,轻混,置于4 ℃冰浴槽0.5 h 以上。
4.1.2.3.3.3.2 将上述6只微量离心管移到 42 ℃水浴中热击2 min,迅即取出再置于4 ℃冰浴槽0.5 h 以上。
4.1.2.3.3.3.3 向上述6只微量离心管各加入1 mL LB培养液,轻混,置于37 ℃水浴槽1h。
4.1.2.3.3.3.4 将18块固态LB板,分别标上 A2、B2、A4、B4、A5、B5号,每号均有3块板;在超净工作台铺板,每板加入上述菌液 200 μL;待板上菌液不会流动时,翻转平板,置于37℃恒温培养箱8h 以上。
4.1.2.3.3.3.5 计算每块板蓝白斑数量,再算出每号管对应的每个处理的蓝白斑总数及百分比。
4.1.2.4 结果判定
在电泳板上,A1、B1、A3、B3号的谱带没有明显差异,说明长时程过量酶处理对 DNA 没有造成额外的切割。B2、B4、B5号和 A2、A4、A5号的白斑总数与其蓝斑总数相比,若比值小于1/9,则判定样品不含核酸外切酶;若比值大于或等于1/9,则判定样品含有核酸外切酶。
4.1.3制定逆转录酶核酸内切酶活性检测方法。
4.1.3.1 仪器
4.1.3.1.1 电泳仪,备电泳槽。
4.1.3.1.2 分析天平(万分之一)。
4.1.3.1.3 紫外透光分析仪。
4.1.3.2 实验步骤
4.1.3.2.1 pUC19 质粒反应
4.1.3.2.1.1 在测试管里先后加入4μLpUC19 质粒 DNA(1μg/μL)、14μL水、2 μL酶,振荡器混匀,水浴1 h。
4.1.3.2.1.2 在对照管里先后加入4 μL pUC19 质粒 DNA(1 μg/μL)、16 μL水,振荡器混匀,水浴1h。
4.1.3.3 电泳
1%~1.5%琼脂糖凝胶,电泳电压5V/cm~10 V/cm,当溴酚蓝线到达凝胶板 2/3时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照。
4.1.3.4 结果判定
在电泳板上,肉眼观察。若测试管和对照管的谱带一致,则判定样品中不含有内切酶;若不一致,则判定样品中含有内切酶。
4.1.4确定逆转录酶核酸内切酶质量标准及验收指标
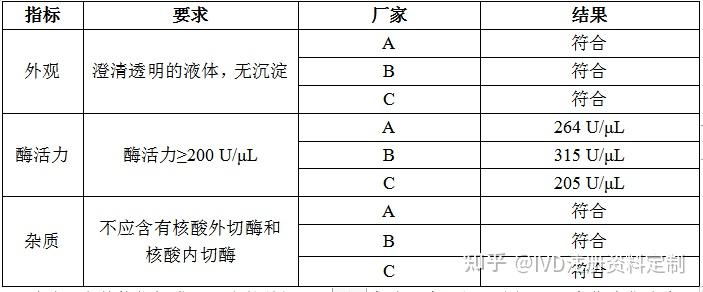
4.1.4.1 外观
澄清透明的液体,无沉淀。
4.1.4.2 酶活力
按4.1.1的要求检查,酶活力≥200 U/μL。
4.1.4.3 杂质
按4.1.2和4.1.3的要求检查,不应含有核酸外切酶和核酸内切酶。
4.1.5逆转录酶核酸内切酶的选择
按上述要求检测结果如下:
结论:在其他指标满足要求的前提下,B厂家酶活力更好,选择B厂家作为供应商,A厂家作为备用供应商。
4.2 TaqDNA聚合酶的选择及质量标准的制定
4.2.1
制定TaqDNA聚合酶活性的检测方法
A.1 仪器设备
全自动医用PCR 分析系统。
A.2 试剂
A.2.1 发夹型寡核苷酸序列(T2),序列为:
5'-tagcgaaggatgtgaacctaatcccTGCTCCCGCGGCCGatctgcCGGCCGCGGGAGCA-3'
A.2.2 dNTP。
注:包括 dCTP、dATP、dTTP、dGTP。
A.2.3 PicoGreen 染料。
A.2.4 Lambda DNA标准品。
A.3 试剂溶液配制
A.3.1 1×TE溶液:10mmol/L Tris-HCl,1 mmol/L EDTA·2Na,pH 8.5(25 ℃)。
A.3.2 PCR buffer:250 mmol/L Tris-HCl,50 mmol/L(NH4)2SO4,500 mmol/L KCl,1%(体积分数)Tritonx-100,pH8.5(25℃)。
A.3.3 Lambda DNA预染液:以8.5 μL:1 μL:0.5 μL 的比例分别量取纯化水、10×PCR buffer 和PicoGreen染料加入EP管;在漩涡混匀器上振荡混匀1 min后在微型离心机上离心数秒.储存干 2℃~8℃。
A.3.4 MgCl2。溶液:25 mmol/L MgCl2·6H2O。
A.3.5 酶稀释缓冲液:10 mmol/L Tris-HCl,0.1 mmol/L EDTA·2Na,1 mmol/L DTT.50 %(体积分数)甘油.pH8.0(25℃)。
A.3.6 T2溶液:将发夹型寡核苷酸序列(T2)干粉按照合成单上的说明稀释到100 μmol/L,储存于-20℃。使用时从-20 ℃取出平衡至室温,于漩涡混匀器上混匀 10 s;在微型离心机上离心数秒。
A.3.7 dNTP混合液:将dNTP中的 dCTP、dATP、dTTP、dGTP等体积进行混合,于漩涡混匀器上混匀 10 s,在微型离心机上离心数秒,配制成浓度为 25 mmol/L的 dNTP混合液。
A.3.8 Lambda DNA标准品溶液:使用1×TE溶液将 Lambda DNA标准品进行 4倍连续梯度稀释,得到 100 μg/mL、25 μg/mL、12.5 μg/mL、6.25 μg/mL、3.125 μg/mL、1.562 5 μg/mL 和0.781 25 μg/mL 8个浓度梯度溶液,以相同体积的1×TE溶液作为背景对照。各梯度溶液和背景对照分别加入等体积的预染液,于漩涡混匀器上混匀10 s,在微型离心机上离心数秒,以 20 μL/管分装于PCR八联管内,每个梯度3个重复,盖紧管盖,在微型离心机上离心数秒后备用。
A.3.9 Taq DNA聚合酶梯度稀释溶液:根据标定的酶比活力,使用酶稀释缓冲液对 Taq DNA聚合酶进行适当稀释(如标定浓度为5000 U/mL,即5U/uL。则可 2倍稀释,得到2×、4×,8×,16×的梯度稀释液)。该步骤应在冰上进行。
A.4 实验步骤
A.4.1聚合反应液的配制与分装
A.4.1.1在 EP管中按照表 2 的比例配制聚合反应液(冰上配制)聚合反应液配制完成后;在漩涡混匀器涡旋混匀10 s,微型离心机离心数秒。
注∶配制总体积计算见式(2):
总体积 =[3×(n+1)+1]×V.......…(A.1)
式中∶
3 —重复次数;
n —Tag DNA聚合酶稀释梯度;
1 —前一个"1"表示空白对照数.后一个"1"表示预留损失数量;
V1—1个反应用量,见表2。
表2聚合反应液配比
A.4.1.2 将A.4.1.1中的聚合反应液平均分装到n个(n为 Taq DNA的稀释梯度数)EP管中,分别加入 1.3 μL 的各浓度梯度的 Taq DNA聚合酶梯度稀释溶液和空白对照(酶稀释缓冲液),在漩涡混匀器涡旋混匀10 s,微型离心机离心数秒。各管混合液以20 μL/管,分装于PCR八联管内,每管混合液3个重复,盖紧管盖,在微型离心机上离心数秒后备用。
A.4.2 运行程序
在全自动医用PCR分析系统上设置如下温度程序∶
74 ℃ 6 s
荧光采集通道∶SYBR
120 cycles
将装有Lambda DNA标准品和聚合反应液的 PCR八联管置于全自动 PCR分析系统中,启动温度程序,开始聚合反应。
A.5 数据分析
A.5.1 Lambda DNA标准曲线的制作
使用全自动医用 PCR分析系统的软件打开数据,导出 Excel文件。
选取Excel文件中Lambda DNA 标准品和背景对照的第 30cycles 的荧光值。计算背景对照3个重复的平均荧光值,将各梯度Lambda DNA标准品的荧光值减掉背景对照的平均荧光值,得到净荧光值。计算各梯度净荧光值的平均值和标准偏差 SD。以Lambda DNA 标准品 DNA量(ng)为 X轴,以各梯度净荧光值的平均值为Y轴,标准偏差 SD为Error 值进行多项式拟合分析.制作工作曲线,从中获得净荧光值与双链 DNA浓度的关系曲线如下,R2≥0.98:
y =A+Bx……(A.2)
式中∶
y——荧光值;
x——双链DNA的量,单位为纳克(ng)。
A.5.2 聚合反应新生成的双链 DNA量的计算
选取Excel文件中待检 Taq DNA聚合酶的第30 cycles 的荧光数据。首先计算空白对照3个重复的平均荧光值,将各梯度 Taq DNA 聚合酶的荧光值减掉空白对照的平均荧光值,得到净荧光值.计算各梯度净荧光值的平均值(y1)。将各净荧光值代入式(A.1)计算聚合反应新生成的双链 DNA量(x1)。
A.5.3 各 Taq DNA 聚合酶梯度条件下的 dNTP消耗量计算
各 Taq DNA 聚合酶梯度条件下的 dNTP消耗量以y。表示,单位为纳摩尔(nmol).按式(A.3)计算∶
y2=x1/(2×324.5)……(A.3)
式中∶
x1——新生成的双链DNA 量.单位纳克(ng)
2 —双链DNA由2 条单链 DNA组成;
324.5 —dNTP的相对平均分子质量。
A.5.4 Taq DNA聚合酶活计算
Tag DNA聚合酶酶活以S表示,单位为 U/mL。选择在0.024 nmol~0.166 nmol范围内的 dNTP 消耗量按照式(A.4)计算∶
S=(y2+0.03)×D/0.24……(A.4)
式中∶
y2——dNTP消耗量,单位纳摩尔(nmol);
D——酶的稀释倍数。
4.2.2
制定TaqDNA聚合酶核酸外切酶活性的检测方法
B.1 原理
cccpUC19 所含经 BamH I位点居于编码Lac Z的α互补肽上,cccpUC19经 BamH I酶切后,由环形 DNA结构形成稳定的具有至黏性末端的双链 DNA.其末端突出部分由五个碱基形成单链.极易受核酸外切酶作用。若样品存在核酸外切酶时,黏性末端易受外切酶切割,再经 T4DNA 连接酶连接,其电泳条带位置不一致。
B.2 仪器和设备
B.2.1 恒温水浴槽。
B.2.2 台式离心机。
B.2.3 超净工作台。
B.2.4 电泳仪。
B.3 材料
B.3.1 高纯ccc pUC19 DNA。
B.3.2 T4 DNA连接酶。
B.3.3 氯仿-异戊醇(24:1,体积比)。
B.3.4 琼脂糖。
B.4 实验步骤
B.4.1 长时程过量酶切
各取5 μg 高纯 ccc pUC19 DNA 置于两只 eppondorf 管,分别标记阴性对照和测试管,在阴性对照管里加入 10 μL 10×BamH I缓冲液.2 μL 限制性内切酶 BamH I(10 U~20 U),加入纯化水至100 μL;在测试管里加入 10 μL 10×BamH Ⅰ缓冲液,2 μL 限制性内切酶 BamH I(10 U~20 U),5 μL TaqDNA聚合酶样品(50 U~100 U),加入纯化水至 100 μL。混匀,37 ℃水浴 10 h,各取出20 μL,分别标记1,2号,待电泳检测;再各取20 μL,分别标记3,4号。
B.4.2 电泳
B.4.2.1 取出1号和2号管,加入载样缓冲液,准备电泳检测。
B.4.2.2 1%~1.5%琼脂糖,电冰电压5 V/cm~10 V/cm;当浪酚蓝线到达凝胶板 2/3时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照。
B.4.3 连接
B.4.3.1 回收 DNA
取出3、4号管,各加入 20 μL氯仿-异戊醇先脱去杂蛋白,再沉淀出 DNA,并真空干燥样品。
B.4.3.2 连接反应
每管加入2 μL 10×T4 DNA连接酶缓冲液,2 U T4 DNA连接酶,加入纯化水至20 uL,混匀,15℃水浴8 h。
B.4.3.3 电泳
B.4.3.3.1 取出3号和 4号管,加入载样缓冲液,准备电泳检测。
B.4.3.3.2 1%~1.5%琼脂糖;电泳电压5V/cm~10 V/cm;当溴酚蓝线到达凝胶板 2/3时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照。
B.5 结果判定
在电泳板上,肉眼观察。若1号和2号的谱带一致,3号和4号的谱带一致,则判定样品中不含核酸外切酶;若1号和2号的谱带不一致或(和)3号和4号的谱带不一致.则判定样品中含核酸外切酶。
4.2.3
制定TaqDNA聚合酶核酸内切酶活性的检测方法
C.1 原理
pUC19质粒为环状结构,核酸内切酶能破坏环状结构,两者电泳谱带不一致。
C.2 仪器
C.2.1 电泳仪。
C.2.2 分析天平(万分之一)。
C.2.3 紫外透光分析仪。
C.3 实验步骤
C.3.1 pUC19 质粒反应
C.3.1.1 在测试管里先后加入4 μLpUC19质粒DNA(1μg/μL)、14 μL纯化水、2 μL 酶,振荡器混匀,水浴1h;
C.3.1.2 在对照管里先后加入4 μL pUC19质粒 DNA(1 μg/μL)、16 μL 纯化水,振荡器混匀,水浴1 h。
C.3.2 电泳
1%~1.5%琼脂糖凝胶,电泳电压5 V/cm~10 V/cm,当溴酚蓝线到达凝胶板 2/3时停止电泳,取出凝胶板置于紫外透光分析仪观察和拍照。
C.4 结果判定
在电泳板上,肉眼观察。若测试管和对照管的谱带一致,则判定样品中不含核酸内切酶;若不一致,则判定样品中含有核酸内切酶。
4.2.4确定TaqDNA聚合酶质量标准及验收指标
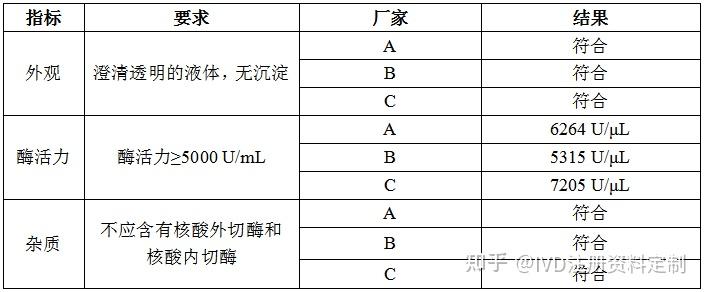
4.2.4.1 外观
澄清透明的液体,无沉淀。
4.2.4.2 酶活力
按4.2.1的要求检查,酶活力≥200 U/μL。
4.2.4.3 杂质
按4.2.2和4.2.3的要求检查,不应含有核酸外切酶和核酸内切酶。
4.2.5TaqDNA聚合酶的选择
按上述要求检测结果如下:
结论:在其他指标满足要求的前提下,C厂家酶活力更好,选择C 厂家作为供应商,A厂家作为备用供应商。
4.3
DNA引物及探针的选择及质量标准的制定
4.3.1 制定DNA引物及探针的检测方法
除非另有说明,在分析中仅使用分析纯试剂,水为 GB/T 6682规定的一级水。
4.3.1.1 外观及性状
感官判断。
4.3.1.2 合成 DNA 的总量检测
4.3.1.2.1 测定吸光度
利用紫外分光光度计测定合成DNA产品的 OD,利用吸光度 A 等于1为1OD单位来计算合成DNA产品的总量。重复测定3次,结果取平均值。
4.3.1.2.2 试验方法
按照GB/T30988执行。
4.3.1.3 核酸引物探针相对分子质量的检测
4.3.1.3.1 相对分子质量的直接计算
相对分子质量可根据引物探针合成单.按照下式直接计算。
MW=(A×313.21)+(C×289.18)+(G×329.21)+(T×304.19)-61.94
式中∶MW
—相对分子质量;
A、C、G、T—各碱基对应的碱基数。
注:计算探针相对分子质量时,应加上相应的修饰基团相对分子质量。常见修饰基团相对分子质量参见GB/T34797表A.1。
4.3.1.3.2 质谱测定相对分子质量
采用ESI源电离喷雾技术,负离子模式将样品转化为运动的带电气态离子碎片,按质核比(m/z)大小分离并记录,通过换算测得相对分子质量。HPLC和 MS的设备及流动相参数参见GB/T34797附录 B。
4.3.1.3.3 测定结果
采用4.3.1.3.2方法测得的相对分子质量与4.3.1.3.1中计算的相对分子质量相对误差应小于或等于0.05%。
4.3.1.4 碱基准确性检测
产品序列和定制引物或定制探针序列的匹配程度,此序列由合成仪后台自动生成并验证。
4.3.1.5 碱基缺失率
通过质谱联用仪对合成的寡核苷酸序列进行测定,计算扣除目标峰以外的其他峰高占所有峰高的比值。
4.3.1.6 引物探针的纯度检测
4.3.1.6.1 质谱法
采用6.3.2 的方法测定,计算目标峰面积占所有峰面积比值得出纯度。
4.3.1.6.2 高效液相色谱法
采用高效液相色谱进行纯度测定,具体条件参见GB/T34797附录B,计算峰面积,并与对照进行比较,得出引物探针纯度。纯度应符合表3的要求。
4.3.1.7 探针修饰基团的准确性
4.3.1.7.1 质谱法
采用6.3.2的方法测定,通过比较相对分子质量来确定是否含有定制探针的修饰基团。
4.3.1.7.2 荧光光谱法
采用荧光光谱仪,固定激发波长,并以激发波长为起始点对发射波长进行扫描(起始点至 900 nm),验证修饰基团的准确性。对应修饰基团的特征吸收峰参见GB/T34797表C.1。
4.3.1.8 引物探针验证
4.3.1.8.1 总则
选择目标产品 DNA作为阳性对照,以非目标产品 DNA作为阴性对照:以灭菌去离子水为空白对照,进行 PCR 扩增(或实时荧光 PCR)试验,根据扩增结果判断试验效果。特异性验证试验应符合GB/T19495.1 的要求。
4.3.1.8.2 特异性和符合性判断
根据扩增图谱查看是否获得特异性条带(或是否有典型扩增曲线)。获得特异性条带(或有典型扩增曲线),即可判断产品符合要求,引物设计合理。
4.3.1.8.3 引物二聚体带
观察 PCR 扩增产物图谱未形成明显的引物二聚体带,空白对照和阴性对照未出现扩增现象,即可判断产品质量合格,引物设计合理。
4.3.4确定引物及探针质量标准及验收指标
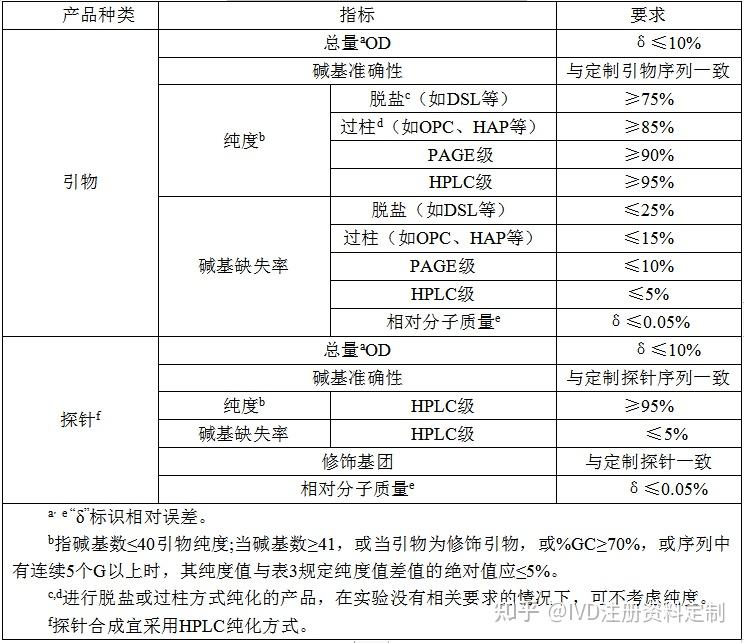
4.3.4.1DNA引物探针产品质量控制指标及技术要求应符合表3的规定。
表3 DNA引物探针产品质量技术要求
4.3.4.2 外观及性状
产品应为半透明或不透明的片状粉状物质、无异味,易溶于水和 TE 缓冲液。
4.3.4.3 引物探针的总量
引物探针的总量一般以OD来表示,合成产品OD测量值与定制序列OD值作比较,要求总量相对误差≤10%。
4.3.4.4 引物探针的相对分子质量
测得的引物探针相对分子质量(MW)与定制序列 MW 值作比较;要求相对分子质量相对误≤0.05%。
4.3.4.5 碱基准确性(DNA序列的一致性)
将合成的引物探针序列与定制序列进行比对,匹配度应达到100%。
4.3.4.6 碱基缺失率
合成产品碱基缺失率应符合表3要求。
4.3.4.7 引物探针的纯度
引物探针纯度主要级别分为"脱盐"级、"过柱"级、PAGE 级、HPLC级,应满足表3要求。
4.3.4.8 探针修饰基团的准确性
探针修饰基团定制序列应完全一致。
4.3.4.9 试验效果评估
将合成以后的引物探针进行PCR扩增以对其试验效果进行评估,应有特异性条带(或有典型扩增曲线),观察 PCR扩增产物图谱未形成明显的引物二聚体带,空白对照和阴性对照未出现扩增现象,即可判断产品质量合格,引物设计合理。
4.3.5DNA引物及探针的选择
4.3.5.1审查供应商合成报告单,满足要求。
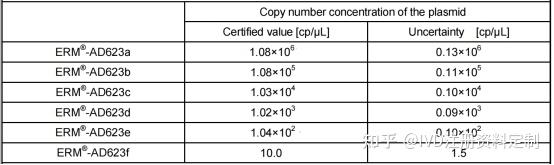
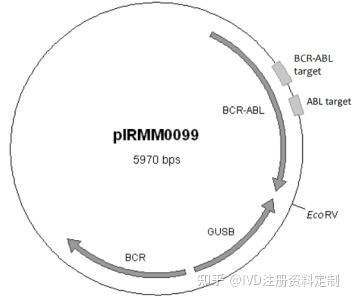
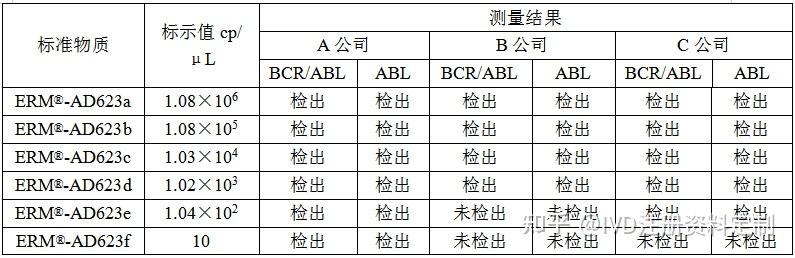
4.3.5.2用欧盟Certified Reference Materials:ERM®-AD623a, ERM®-AD623b,ERM®-AD623c,ERM®-AD623d, ERM®-AD623e,ERM®-AD623f(plasmid DNA containing a BCR-ABL b3a2 transcript fragment)进行验证。
标准物质浓度如下:
质粒图谱如下:
结论:在其他指标满足要求的前提下,A厂家灵敏度更高,选择A 厂家作为供应商。
4.4质控品原料选择及质量标准的制定
4.4.1实验目的:确定合适的BCR/ABL和ABL细胞系。
4.4.2 试验仪器
仪器:离心机、移液器、低温高速离心机、恒温水浴锅、数字 PCR 仪(型号:QX200 droplet reader, IVD),BIO-RAD公司生产;
4.4.3 耗材与试剂
耗材:1000ul枪头、200ul枪头、10ul枪头、EP管。
试剂:美国Bio-Rad的BCR-ABL数字PCR法试剂盒(QXDx BCR-ABL %IS Kit),不同厂家的K562细胞系,HL60细胞系。
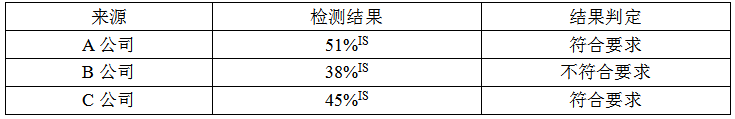
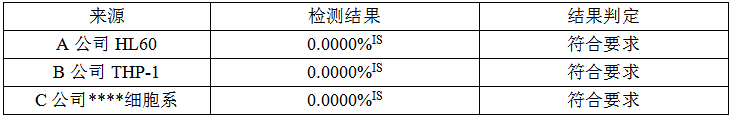
4.4.4质量标准:K562细胞系融合基因浓度:>40%IS,HL60细胞系细胞系融合基因浓度:<0.002%IS。
4.4.4 实验步骤
将K562细胞系、KL60细胞系按试剂盒的要求提取RNA,反转录,数字PCR反应并进行数据分析结果如下:
K562细胞系检测结果如下:
BCR-ABL阴性细胞系检测结果如下:
结论:从结果看,A、C公司的K562细胞系、KL60细胞系均满足质量标准,由于A公司K562细胞系融合基因mRNA浓度最高,将其作为原料供应商。
五、 原材料研究总结
综上所述:
反转录酶选择选择****公司;引物和探针选择****公司;TapDNA聚合酶选择****公司;K562细胞系、KL60细胞系选择****有限公司。
详细原材料清单请见附件1。
附件1: 原材料清单
序号
原辅料名称
1
逆转录酶(c-MMLV酶)
2
反转录酶
3
RNasin
4
逆转录引物:随机六核苷酸引物
5
热启动TaqDNA聚合酶
6
BCR-ABL 引物和探针
7
ABL 引物和探针
8
去核酸酶水
9
K562 cells
10
HL60 cells
其他医疗器械/体外诊断试剂注册资料模板参见:
【墨斗鱼平台】注册资料分享 - 百度文库
原文地址:https://zhuanlan.zhihu.com/p/451156691
回复
举报
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
浏览过的版块
行业研究
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X5.0 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-18 05:19
发表于 2025-3-18 05:19