同一时期,Macosko等发明了类似的 Slide-Seq 方法。在Slide-seq中,首先在涂有橡胶的玻璃盖玻片表面填充一层10 μm的特异性条形码微珠(beads)。与其他高通量scRNA-seq方法中使用的微珠类似,每个微珠上条形码是随机分布的。因此,先对每个微珠上寡核苷酸条形码进行识别,再进行混合测序。为解决这一问题,Slide-seq后续利用SOLiD化学技术(sequencing by oligonucleotide ligation and detection)来读取每个柱子上不同条形码。先识别每个10 μm捕获点(spot)中特异性空间标识符,再行RNA-seq,因此寡核苷酸捕获的mRNAs可以映射回它们的原始空间坐标。
ISS 技术中,锁式探针(padlock probe)可能会产生大量的探针特定偏差,而且 ISS 技术通常仅限于有明确注释基因的检测。针对这些问题,2015 年,Lee 等开发了荧光原位测序(fluorescence in situ sequencing,FISSEQ)技术,利用随机六聚体引物在固定细胞中逆转录 RNA,通过 cDNA 的环化以非靶向方式生成测序文库。进一步提高了原位测序技术的检测通量,能获得全基因组范围的基因表达图谱,包括基因表达,RNA 剪接和转录后修饰等,同时保留了空间位置信息。
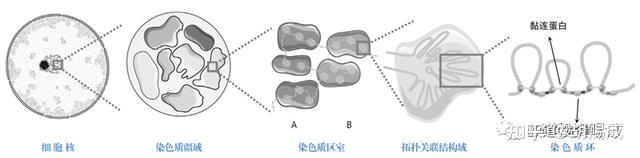
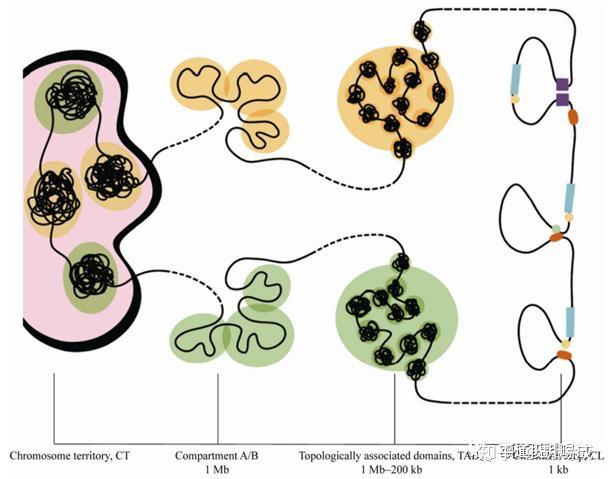
随着测序精度的提升,研究人员发现区室内部 1Mb 左右的 DNA 组成了更小的空间结构,称为拓扑关联结构域(topologically associating domain, TAD),TAD内一般包含 8~10 个基因,其内部的 DNA 元件之间形成了较为紧密的相互作用,而 TAD 间的染色质相互作用较少。相邻 TAD 边界上结合有染色质结构蛋白,如CTCF 蛋白(CCCTC binding factor, CTCF)和黏连蛋白的蛋白复合体,这些蛋白起到组织染色质结构并隔离两个相邻的 TAD 之间互作的功能。
TADs 参与调控 DNA 复制、转录和表观遗传修饰,因此,TAD 边界的破坏会对基因表达产生较大规模的影响,甚至导致疾病的发生。通过技术分析TADs的变化是否与表观遗传修饰相关,结合RNA-seq 技术对相关基因的表达量进行统计,有助于解释不同样本间空间结构的差异与表观遗传修饰及转录调控之间的关系。
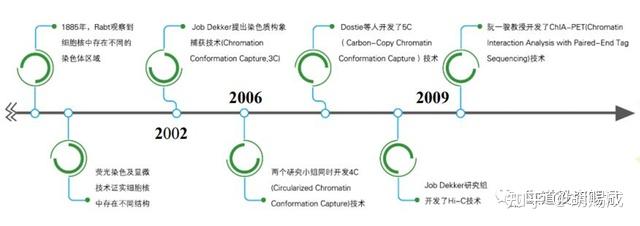
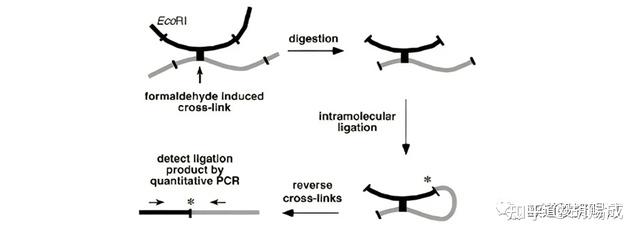
4C 技术与 3C 技术相比省略了 PCR 步骤,直接用限制酶切割染色质的目标 DNA 并环化,然后利用反向 PCR 技术对目标染色质位点接触的所有 DNA 序列进行扩增,最后利用微阵列分析或下一代测序技术(NGS)分析某一特定 DNA 序列与其他接触位点DNA 序列的相互作用。该技术可以在全基因组范围内检测与靶向基因座接触的 DNA 基因座,许多线性距离较远的染色质间的相互作用也可通过形成染色质接触而被检测出来。
Hi-C技术技术是 3C 技术的一个高通量版本,能够检测所有目标基因组位点的所有相互作用。它的研究对象为整个细胞,通过捕获细胞内染色质全部 DNA 的相互作用模式,从而研究整个染色质 DNA 在空间位置上的关系,还可以进一步得到分辨率较高的染色质三维结构信息。
Hi-C 技术改变了 3C 技术中创立模板的过程,在连接目标 DNA 末端之前,用生物素标记的脱氧核苷酸填充 DNA 的限制性末端。在 DNA 末端连接之后,将其纯化并剪切,然后富集被亲和素标记的连接头部分进行分析。最后得到整个基因组 DNA 片段之间的相互作用频率矩阵,其分辨率与限制性位点的密度以及测序深度相关。

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-1-18 12:09
发表于 2025-1-18 12:09



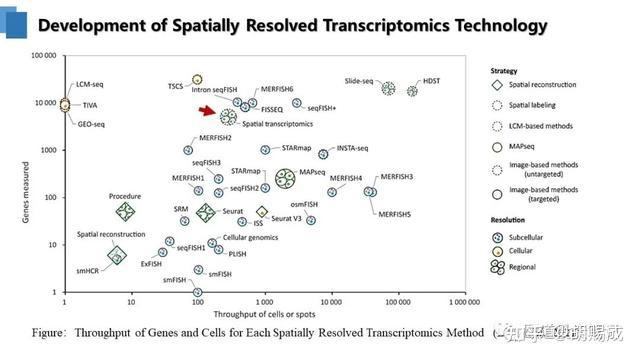
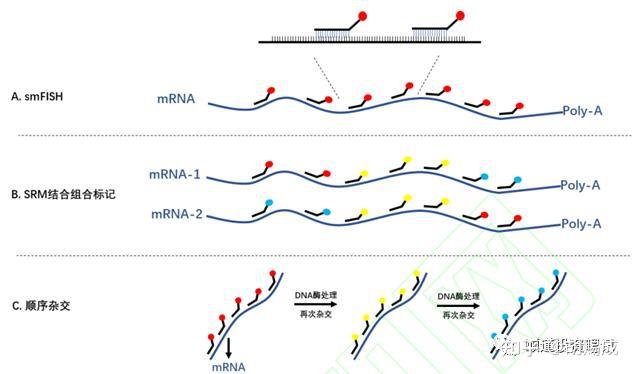
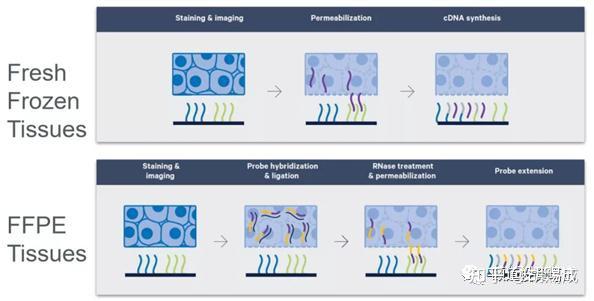
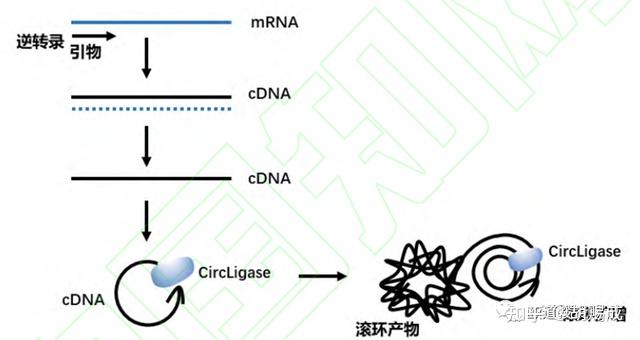

 提升卡
提升卡


