|
摘要:即将实施的《医疗器械注册管理办法》及《体外诊断试剂注册管理办法》对“延续注册”进行了全面简化。那么应如何实现不影响销售的成功延续?何时启动延续注册项目? 何时开始申请证书延续?——在医疗器械注册证有效期届满6个月前提出申请。 即将于2014年10月1日实施的《医疗器械注册管理办法》(以下简称“《办法》”)的第五十四条规定:“医疗器械注册证有效期届满需要延续注册的,注册人应当在医疗器械注册证有效期届满6个月前,向食品药品监督管理部门申请延续注册,并按照相关要求提交申报资料。”第五十五条同时指出:“注册人未在规定期限内提出延续注册申请的,不予延续注册”具体如下图: 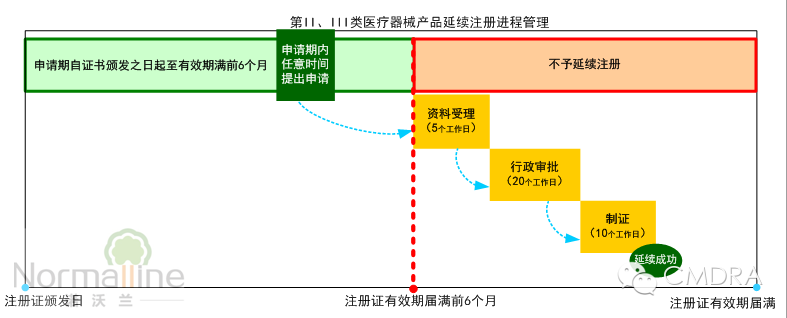
从上图可以看出,简化后的延续注册审评审批时限仅35个工作日,比现行的101个工作日大幅缩短。《办法》同时规定“监督管理部门应当在医疗器械注册证有效期届满前作出准予延续的决定,逾期未作决定的,视为准予延续。”也就是说只要申请人在《办法》规定期限内提出延续注册申请,几乎不可能出现“原注册证已过期失效而新注册证还未获准,处于无证的状况”的情况。 再来看一下将导至申请人无法在6个月内完成证书延续的“例外情况”。较常见的有:由于产品本身的变化以及相关技术标准或规定的变化,使得产品无法符合延续注册的规定,而必须申请许可事项变更注册。如更换医疗器械产品(或体外诊断试剂)的主要原材料供应商;改变骨科植入物的表面喷砂处理工艺;升级有源器械的软件都属于产品的实质性变更,依据规定必须申请许可事项变更。 较之延续注册,许可事项变更通常要求对产品变化进行安全性有效性的再评价,因此涉及更复杂的验证过程(如注册检测)及资料。并且许可事项变更的审评审批时间也较长(第II类产品通常为98个工作日,第III类产品通常为128个工作日)。因此许可事项变更很少能在6个月内完成。 依据相关规定,未获得注册证书的医疗器械不得销售、使用,势必影响企业产品的销售。因此在业内常将“不影响销售”作为注册证书成功延续的考量指标。所以为实现“不影响销售的证书延续”的目标,通常至少提前12个月而不是6个月,启动延续注册申请项目,评估适合企业的申请途径。
本文转自CMDR | 
 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2014-8-1 13:42
发表于 2014-8-1 13:42


 提升卡
提升卡 发表于 2014-8-2 21:33
发表于 2014-8-2 21:33



